9月11日出版的《Nature Genetics》刊载了加州大学圣地亚哥分校(UCSD)生物信息学的研究人员的最新研究。他们联合纽约基因组中心、哈佛大学和麻省理工学院共同开发了一种可分析人类基因组中短串联重复序列(short tandem repeats,STR)或称为微卫星(microsatellites)突变的工具。
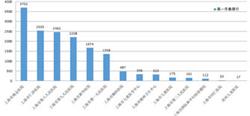
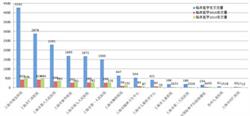
研究人员详细描述了他们所构建的数学模型,以此来预测人类基因组中重复序列的出现和突变频率。他们从300个独立个体基因组中获取了超过150万个重复序列。
在他们早期开发的能精确估计Y染色体中串联重复序列的MUTEA算法基础上,经改进后,这款新算法可用于分析成对的DNA变异(haplotypes,单倍型)。采用新算法分析后,研究人员发现,不同种类的突变不仅有规律可循,而且间隔时间可被预测,因此他们构建了所谓的“分子钟”。利用分子钟,可确定突变在基因组内的突变频率。
研究人员使用模型计算实际突变率,并与预期突变率进行比较。基因组中在生命早期产生严重健康状况的突变往往比预期的突变少,遗传学家将这种情况定义为高度被约束的。例如,患有自闭症的人不太可能会将他们的基因遗传给下一代。相反,例如亨廷顿氏病等导致生命后期发病的突变很可能会传给孩子,通常不受约束。
研究人员对一些早发型和晚发型疾病的重复序列套用了他们的模型。该模型正确地鉴定出了受约束的与早发条件有关的重复序列。
此外,他们还利用这个方法分析了一组非疾病相关重复序列,结果与预期一致,这些重复序列按照预期的速率发生变化并且不受约束。
原文链接:http://www.nature.com/ng/journal/vaop/ncurrent/full/ng.3952.html?foxtrotcallback=true