碱基编辑是一项具有巨大潜力的基因治疗技术,它可以用来修复或修改个体的基因,从而治疗一些遗传性疾病和其他疾病。碱基编辑器可以在不产生双链DNA断裂的基础上高效催化碱基转换,主要包括ABE和CBE两种,分别实现A-to-G和C-to-T的转换。然而,研究发现gRNA在靶向编辑的同时会与非靶点DNA序列错配,引入非预期的基因突变,即脱靶编辑,这严重制约了基因编辑技术的广泛应用。传统的检测脱靶实验既耗时又成本高。为了解决这个问题,2023年9月2日,公共卫生学院王慧课题组联合复旦大学生命科学学院王永明课题组、复旦大学附属浦东医院余波课题组在Nature Communications上发表了题为《基于深度学习的碱基编辑脱靶预测》(Prediction of base editor off-targets by deep learning)的文章,该研究的共同第一作者为公共卫生学院的张成东助理研究员及其团队成员杨元。该研究利用深度学习的方法针对ABE和CBE分别构建了gRNA脱靶的预测模型,为它们的广泛应用奠定了基础。这些模型可以在网站上免费在线使用(http://www.deephf.com/#/bedeep/bedeepoff),也可以通过代码仓库在本地部署使用(https://github.com/izhangcd/BEdeep)。

首先,研究者针对ABE和CBE分别设计了超过9万对gRNA和脱靶序列,这些序列覆盖了1~6bp的错配和1~2bp的插入缺失等脱靶类型。通过将每个gRNA及其脱靶序列合成到一个oligo DNA上建立慢病毒文库。将文库感染表达ABE和CBE的细胞,待脱靶序列编辑后,使用PCR扩增并进行高通量测序,最终得到每个gRNA的脱靶编辑效率。脱靶编辑效率被定义为包含特定碱基编辑类型的Reads数量与总Reads数量的比值。该研究过滤总Reads数量少于100的gRNA-脱靶序列组合,以获得高质量的数据集。为消除不同gRNA靶向编辑效率的差异,使用脱靶与靶向编辑效率的比值代替原始的脱靶编辑效率,用以表示对脱靶编辑的耐受情况。脱靶类型分析发现所有的突变类型都会导致编辑效率比值降低。此外,脱靶位置分析发现1~10位的突变比11~20位的突变对编辑效率比值的影响更小。
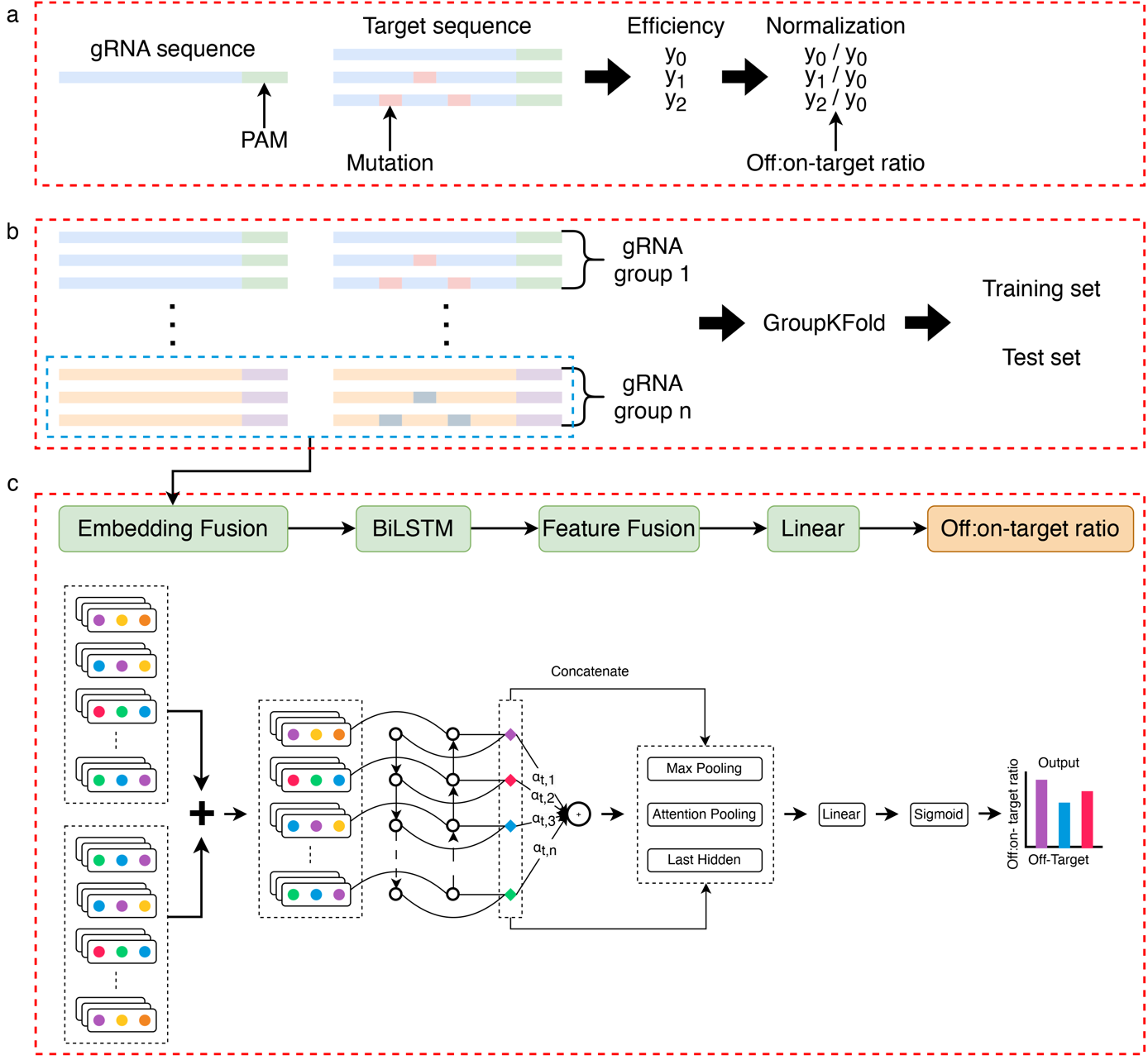
图1.BEdeepoff流程图a.脱靶编辑效率的标准化。b.数据集拆分得到训练集和测试集。c.深度学习模型结构示意图
其次,研究者训练了一种基于融合嵌入的深度学习模型,包含Embedding,LSTM,Attention等模块。模型将gRNA和脱靶序列作为输入,多种脱靶序列共享相同的gRNA。为防止训练过程中gRNA的信息泄漏,根据gRNA序列将gRNA和脱靶序列对分组,并使用"GroupKFold"的方式将数据集分为训练集和测试集。内部测试数据集的结果表明模型在1~2 bp的错配和1bp的插入缺失上性能表现较好。对于外部内源位点测试数据,模型预测结果与真实值之间的相关性在0.710-0.859。特征归因分析发现突变位置的归因分数都在0以下,表明这些位置对最终的预测结果具有负贡献。
该研究通过结合高通量测序与深度学习实现了ABE和CBE的脱靶预测,模型结构简单高效,填补了碱基编辑脱靶预测问题的空白,有助于帮助研究者使用设计良好的RNA引导序列,降低脱靶可能性。同时该方法也可以推广至其他碱基编辑器的脱靶预测。
复旦大学生命科学学院王永明教授、复旦大学附属浦东医院余波教授、公共卫生学院/单细胞组学与疾病研究中心王慧教授为该论文的通讯作者。公共卫生学院/单细胞组学与疾病研究中心张成东助理研究员、杨元硕士为该论文共同第一作者。该研究得到科技部、基金委和上海市科委等多项基金的资助。
原文链接:https://doi.org/10.1038/s41467-023-41004-3
