12月18日,国际学术期刊Cardiovascular
Research在线发表了健康科学研究所杨黄恬研究组题为“Uncoupling protein 3 mediates H2O2
preconditioning-afforded cardioprotection through the inhibition of
MPTP opening”
的最新研究成果,该研究揭示了H2O2预处理(H2O2PC)和UCP3的心肌保护作用及其线粒体调控的分子机制。
心肌梗塞后及时再灌注是挽救缺血心肌必需的步骤,但该过程伴随着严重的再灌注损伤。缺血/再灌注造成的线粒体Ca2+([Ca2+]m)超载和活性氧(reactive
oxygen
species,ROS)大量释放并导致线粒体严重的结构和功能破坏,是心肌细胞缺血/再灌注损伤的主要原因。因此寻找保护线粒体的关键靶点,对于深入理解缺血/再灌注导致心肌细胞结构和功能损伤机制,进而发展心肌保护的新策略至关重要。ROS不仅参与细胞损伤,适量ROS
通过激活细胞保护信号通路介导缺血预处理/后处理的抗心肌缺血/再灌注损伤的保护作用。线粒体是调控心肌细胞活性氧产生、Ca2+稳态和收缩功能的重要细胞器之一。在缺血/再灌注过程中过度释放的活性氧以及线粒体Ca2+
超载都被证明可以打开线粒体膜通透性通道(mitochondrial permeability transition pores,
MPTP),进而造成心肌细胞的凋亡和坏死。因此理解和阐明在缺血/再灌注过程中线粒体ROS
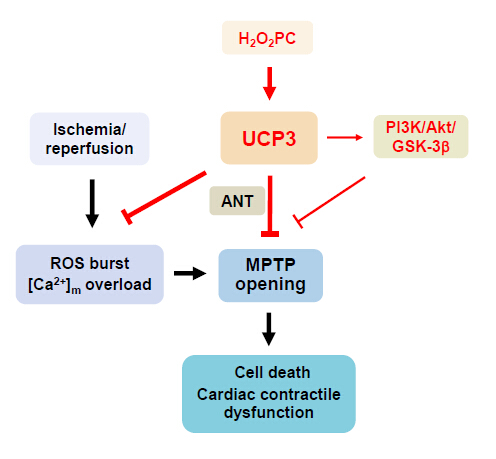
大量释放与Ca2+超载的发生原因以及MPTP开放的分子调控机制对于发展有效的心肌保护措施至关重要的。线粒体解偶联蛋白(mitochondrial
uncoupling proteins,UCPs)位于线粒体内膜,可抵抗心脏缺血/再灌注损伤,但确切机制尚不清楚。
博士研究生陈意雄等在杨黄恬研究员的指导下发现H2O2PC显著升高心肌内UCP3的表达,而UCP3介导和模拟H2O2PC在缺血/再灌注过程中改善心功能、降低心肌梗死面积、抑制[Ca2+]m超载和线粒体ROS大量释放及防止ΔΨm
丧失。H2O2PC和UCP3 过表达一方面通过UCP3直接与MPTP
通道蛋白ANT相互作用而抑制MPTP的开放起到保护作用,另一方面可以启动保护性信号通路PI3K/Akt,进而抑制GSK-3活性。MPTP在H2O2PC和UCP3的抗心肌缺血/再灌注损伤的保护作用起决定性作用,而PI3K/Akt/GSK-3
仅部分参与H2O2PC和UCP3 的心肌保护作用。
此研究得到了国家自然科学基金委和国家科技部973项目的资助。
论文在线发表网址:
http://cardiovascres.oxfordjournals.org/content/early/2014/12/16/cvr.cvu256



